
多选题关于苯丙酮尿症(PKU)的叙述错误的是()A属常染色体显性遗传B典型PKU系由于患儿肝细胞缺乏苯丙氨酸羟化酶C经饮食控制后,脑电图正常,特殊气味消失D出生后早发现早治疗可预防智能发育落后E尿及汗液中排除苯丙酮酸致使有特殊的鼠尿样臭味
题目
属常染色体显性遗传
典型PKU系由于患儿肝细胞缺乏苯丙氨酸羟化酶
经饮食控制后,脑电图正常,特殊气味消失
出生后早发现早治疗可预防智能发育落后
尿及汗液中排除苯丙酮酸致使有特殊的鼠尿样臭味
相似考题
参考答案和解析
更多“多选题关于苯丙酮尿症(PKU)的叙述错误的是()A属常染色体显性遗传B典型PKU系由于患儿肝细胞缺乏苯丙氨酸羟化酶C经饮食控制后,脑电图正常,特殊气味消失D出生后早发现早治疗可预防智能发育落后E尿及汗液中排除苯丙酮酸致使有特殊的鼠尿样臭味”相关问题
-
第1题:
关于苯丙酮尿症(PKU)的叙述错误的是
A、属常染色体显性遗传
B、典型PKU系由于患儿肝细胞缺乏苯丙氨酸羟化酶
C、经饮食控制后,脑电图正常,特殊气味消失
D、出生后早发现早治疗可预防智能发育落后
E、尿及汗液中排除苯丙酮酸致使有特殊的鼠尿样臭味
参考答案:AE
-
第2题:
男孩,3岁。生后5个月见体格发育落后,表情呆滞,伴有点头、弯腰样发作,每日约10余次。2岁开始出现呕吐,喂养困难。现体检见小儿智力明显落后,毛发棕黄色,肤色浅,尿无明显霉臭味,尿三氯化铁试验出现绿色。脑电图示高峰节律紊乱。该患儿最可能的诊断为 ( )A、苯丙酮尿症
B、婴儿痉挛症
C、半乳糖血症
D、癫痫
E、以上诊断都有可能
上述患儿如为经典型苯丙酮尿症,其发生的最主要机制是 ( )A、肝细胞内苯丙氨酸羟化酶缺乏
B、体内有过多的苯丙酮酸
C、苯丙氨酸在体内蓄积
D、组氨羟化酶被抑制制
E、四氢生物喋呤生成不足
上述患儿如为非经典型苯丙酮尿症,其发生的最主要机制是 ( )A、二氢生物喋呤还原酶先天缺乏
B、高苯丙氨酸血症
C、酪氨羟化酶被抑制制
D、血酪氨酸浓度下降
E、四氢生物喋呤生成不足
该患儿系经典型苯丙酮尿症,其血浆苯丙氨酸浓度范围是 ( )A、0.061~0.18mmol/L
B、0.18~0.36mmol/L
C、0.36~4.88mmol/L
D、4.89~5.55mmol/L
E、>1.22mmol/L
该患儿系经典型苯丙酮尿症,应尽早开始饮食治疗。下列哪项不妥 ( )A、给予低苯丙氨酸饮食,以预防脑损害及智能低下发生
B、严格的饮食控制,主要使用于血浆苯丙氨酸水平持续高于1.22mmol/L者
C、最好采用低苯丙氨酸配方,达到既限制苯丙氨酸摄入有保证正常生长发育之需
D、6个月以后辅食的增加与正常儿相仿,要选择苯丙氨酸低的食品
E、适当控制苯丙氨酸的摄入,持续至成人
参考答案:问题 1 答案:A
问题 2 答案:A
问题 3 答案:E
问题 4 答案:E
问题 5 答案:E
-
第3题:
4岁男孩。生后5个月见表情呆滞,易激惹,不能抬头,伴有点头、弯腰样发作,每日约10余次。2岁开始出现呕吐,喂养困难。现见小儿智能明显落后,毛发棕黄色,皮肤嫩,尿为鼠臭味,尿三氯化铁试验出现绿色。该患儿最可能的诊断是( )A、组氨酸血症
B、苯丙酮尿症
C、半乳糖血症
D、同型胱氨酸尿症
E、高精氨酸血症
上述患儿系经典型苯丙酮尿症,其发生的最主要机制是( )A、体内有过多的苯丙酮酸
B、苯丙氨酸在体内蓄积
C、四氢生物蝶呤生成不足
D、组氨酸羟化酶被抑制
E、肝细胞内苯丙氨酸羟化酶缺乏
假如上述患儿系非经典型苯丙酮尿症,其发生的最主要机制是( )A、高苯丙氨酸血症
B、四氢生物蝶呤生成不足
C、血酪氨酸浓度下降
D、酪氨酸羟化酶被抑制
E、二氢生物蝶呤还原酶先天缺陷
该患儿系经典型苯丙酮尿症,其血浆苯丙氨酸浓度范围是( )A、0.06~0.08mmol/L
B、0.08~0.16mmol/L
C、0.24~1.20mmol/L
D、2.44~5.55mmol/L
E、>5.55mmol/L
该患儿系经典型苯丙酮尿症,应尽早开始饮食控制。下列哪项不妥当( )A、出生后适当控制苯丙氨酸的摄入,持续至成人
B、6个月以后辅食的增加与正常小儿相仿,要选择含苯丙氨酸低的食品
C、最好采用低苯丙氨酸配方,达到既限制苯丙氨酸摄入,又保证正常发育的需要
D、给予低苯丙氨酸饮食,以预防脑损害及智力低下
E、终生适当控制苯丙氨酸的摄入
参考答案:问题 1 答案:B
问题 2 答案:E
问题 3 答案:E
问题 4 答案:C
问题 5 答案:A
-
第4题:
苯丙酮尿症(PKU)的遗传方式是A.常染色体隐性遗传
B.X-连锁显性遗传
C.伴性不完全显性遗传
D.常染色体显性遗传
E.X-连锁隐性遗传答案:A解析: -
第5题:
由于尿液和汗液中排出苯丙酮酸,苯丙酮尿症患儿呈特殊的鼠尿臭味。( )
正确答案:错误 -
第6题:
苯丙酮尿症(PKU)的遗传方式是()
- A、常染色体显性遗传
- B、常染色体隐性遗传
- C、X-连锁显性遗传
- D、X-连锁隐性遗传
- E、伴性不完全显性遗传
正确答案:B -
第7题:
4岁男孩。生后5个月见表情呆滞,易激惹,不能抬头,伴有点头、弯腰样发作,每日约10余次。2岁开始出现呕吐,喂养困难。现见小儿智能明显落后,毛发棕黄色,皮肤嫩,尿为鼠臭味,尿三氯化铁试验出现绿色。 上述患儿系经典型苯丙酮尿症,其发生的最主要机制是()
- A、体内有过多的苯丙酮酸
- B、苯丙氨酸在体内蓄积
- C、四氢生物蝶呤生成不足
- D、组氨酸羟化酶被抑制
- E、肝细胞内苯丙氨酸羟化酶缺乏
正确答案:E -
第8题:
单选题苯丙酮尿症临床表现最突出的是()A行为异常
B智能发育落后
C皮肤和虹膜色浅
D尿及汗液呈鼠尿样臭味
E肌张力减低
正确答案: B解析: 暂无解析 -
第9题:
单选题苯丙酮尿症患儿经治疗最难改善的是()A惊厥
B脑电图
C毛发、皮肤色泽
D智能低下
E尿、汗液特殊气味
正确答案: E解析: 暂无解析 -
第10题:
判断题由于尿液和汗液中排出苯丙酮酸,苯丙酮尿症患儿呈特殊的鼠尿臭味。( )A对
B错
正确答案: 错解析: 暂无解析 -
第11题:
单选题关于典型苯丙酮尿症,错误的是()A常染色体隐性遗传
B肝细胞缺乏苯丙氨酸羟化酶
C四氢生物蝶呤缺乏
D大量苯丙酮酸生成
E甲状腺素、肾上腺素合成减少
正确答案: A解析: 暂无解析 -
第12题:
有关苯丙酮尿症(PKU),叙述错误的是
A、是一种常见的遗传性氨基酸代谢病
B、属常染色体显性遗传病,未经治疗的PKU患儿由于体内蓄积过量的苯丙氨酸及其代谢产物,对神经产生毒性作用,最终出现严重的智能障碍。
C、是由于编码苯丙氨酸羟化酶的基因发生突变,致使PAH活性降低或丧失,苯丙氨酸代谢异常所致
D、中国人群PAH基因突变近30余种
E、可以利用PAH基因中短串联重复序列(如内含子3内TCTA n)扩增片段长度多态性进行连锁分析,产前诊断PKU
参考答案:B
-
第13题:
患儿,1岁。生后4个月发现智力发育落后,尿有鼠尿臭味,毛发及皮肤颜色浅,7月开始出现癫痫小发作。实验室检查确诊为典型的PKU,给予低苯丙氨酸饮食。该患儿每天允许摄入苯丙氨酸的量为
A、5~10mg/kg
B、10~20mg/kg
C、20~30mg/kg
D、30~50mg/kg
E、50~70mg/kg
参考答案:D
-
第14题:
关于苯丙酮尿症描述错误的是( )。A.属于常染色体隐性遗传病,患儿智力发育落后
B.发病与苯丙酮酸羟化酶基因突变导致酶活性降低有关
C.患儿特有体征鼠尿味
D.该病越早治疗越好,低苯丙氨酸饮食治疗至少维持到青春期答案:B解析:苯丙酮尿症的发病机理是苯丙氨酸羟化酶缺陷,使苯丙氨酸和苯丙酮酸在体内堆积而致病。所以题中选项B表述不正确。 -
第15题:
男孩,3岁。生后4个月见表情呆滞,易激惹,不能抬头,伴有惊厥,每日约10余次。2岁开始出现呕吐,喂养困难。现见小儿智能明显落后,毛发棕黄色,皮肤嫩,尿为鼠臭味,尿三氯化铁试验出现绿色,血清T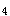 、T
、T 、TSH测定在正常范围。如该患儿系经典型苯丙酮尿症,应尽早开始饮食控制。下列哪项不可采用
、TSH测定在正常范围。如该患儿系经典型苯丙酮尿症,应尽早开始饮食控制。下列哪项不可采用
A.出生适当控制苯丙氨酸的摄入,持续终生
B.采用低苯丙氨酸配方,达到既限制苯丙氨酸摄入,又保证正常发育的需要
C.苯丙氨酸需要量2个月以内约需50~70mg/(kg·d)
D.苯丙氨酸需要量4岁以上约10~30mg/(kg·d)
E.低苯丙氨酸饮食主要适用于典型PKU答案:A解析:苯丙酮酸尿症根据智能落后,头发由黑变黄,特殊体貌,尿三氯化铁试验可确诊。本病发病机制由于患儿肝脏缺乏苯丙氨酸羟化酶活性,导致苯丙氨酸浓度增高。 -
第16题:
苯丙酮尿症患儿的特征性临床表现是()
- A、智能发育落后
- B、惊厥
- C、毛发皮肤色泽变浅
- D、肌张力增高
- E、尿和汗液有鼠尿臭味
正确答案:E -
第17题:
苯丙酮尿症患儿绝大多数为缺乏苯丙氨酸-4-羟化酶所致,是氨基酸代谢障碍中较常见的一种,属常染色体显性遗传。( )
正确答案:错误 -
第18题:
苯丙酮尿症患儿最突出临床表现是()
- A、惊厥
- B、智能发育落后
- C、肌张力增高
- D、毛发、皮肤色泽变浅
- E、尿和汗液有鼠臭味
正确答案:B -
第19题:
多选题关于苯丙酮尿症(PKU)的叙述错误的是()A属常染色体显性遗传
B典型PKU系由于患儿肝细胞缺乏苯丙氨酸羟化酶
C经饮食控制后,脑电图正常,特殊气味消失
D出生后早发现早治疗可预防智能发育落后
E尿及汗液中排除苯丙酮酸致使有特殊的鼠尿样臭味
正确答案: E,B解析: 暂无解析 -
第20题:
单选题苯丙酮尿症的主要诊断依据()A智能发育落后
B尿三氯化铁试验阳性
C尿有鼠尿样臭味
D脑电图异常
E血清苯丙氨酸浓度升高
正确答案: A解析: 暂无解析 -
第21题:
单选题苯丙酮尿症(PKU)的遗传方式是()A常染色体显性遗传
B常染色体隐性遗传
CX-连锁显性遗传
DX-连锁隐性遗传
E伴性不完全显性遗传
正确答案: B解析: 暂无解析